- THERMODYNAMIQUE - Lois fondamentales
- THERMODYNAMIQUE - Lois fondamentalesLe principe d’équivalence des unités de chaleur et de travail est généralement attribué au médecin allemand J. R. von Mayer, qui l’a formulé pour la première fois en 1842 dans ses Remarques sur les forces inanimées de la nature. Mais on doit aussi associer à la même découverte le nom de J. P. Joule avec, comme précurseurs, B. Thompson (comte Rumford, 1753-1814) et, semble-t-il aussi, Sadi Carnot (1796-1832), connu surtout pour sa contribution fondamentale au second principe.1. Le premier principePour un système fermé, le principe d’équivalence conduit à l’expression générale suivante du premier principe (dans un système unifié d’unités):
 La quantité U2 漣 U1 correspond à l’accroissement de l’énergie U du système entre l’état initial 1 et l’état final 2. La quantité Q est la chaleur reçue par le système, et W est le travail fourni au milieu extérieur.L’énergie U se présente donc comme une fonction d’état, et le premier principe de la thermodynamique exprime dans sa généralité une propriété de conservation de cette énergie, car, pour un système isolé du monde extérieur, on a Q = 0 et W = 0, ce qui entraîne U1 = U2. D’où le nom de principe de la conservation de l’énergie , dont l’adoption revient à admettre l’impossibilité d’un moteur perpétuel et exige, en outre, l’abandon du vieux concept de calorique (cf. THERMODYNAMIQUE - Histoire).Il est souvent commode de disposer de la forme différentielle du premier principe en vue de son application aux transformations infinitésimales. On écrit dans ce cas:
La quantité U2 漣 U1 correspond à l’accroissement de l’énergie U du système entre l’état initial 1 et l’état final 2. La quantité Q est la chaleur reçue par le système, et W est le travail fourni au milieu extérieur.L’énergie U se présente donc comme une fonction d’état, et le premier principe de la thermodynamique exprime dans sa généralité une propriété de conservation de cette énergie, car, pour un système isolé du monde extérieur, on a Q = 0 et W = 0, ce qui entraîne U1 = U2. D’où le nom de principe de la conservation de l’énergie , dont l’adoption revient à admettre l’impossibilité d’un moteur perpétuel et exige, en outre, l’abandon du vieux concept de calorique (cf. THERMODYNAMIQUE - Histoire).Il est souvent commode de disposer de la forme différentielle du premier principe en vue de son application aux transformations infinitésimales. On écrit dans ce cas: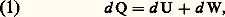 où d Q désigne la quantité de chaleur élémentaire reçue par le système pendant le temps dt , et d W le travail correspondant fourni au milieu extérieur.Il faut cependant souligner que, dans cette formulation, seule la quantité d U représente une différentielle exacte des variables décrivant l’état du système.Dans le cas particulier d’un fluide à pression p uniforme, le travail élémentaire d W se réduit au produit pd V et l’énergie totale U, à l’énergie interne E en l’absence d’énergie cinétique. Le principe de conservation de l’énergie prend alors la forme simplifiée usuelle:
où d Q désigne la quantité de chaleur élémentaire reçue par le système pendant le temps dt , et d W le travail correspondant fourni au milieu extérieur.Il faut cependant souligner que, dans cette formulation, seule la quantité d U représente une différentielle exacte des variables décrivant l’état du système.Dans le cas particulier d’un fluide à pression p uniforme, le travail élémentaire d W se réduit au produit pd V et l’énergie totale U, à l’énergie interne E en l’absence d’énergie cinétique. Le principe de conservation de l’énergie prend alors la forme simplifiée usuelle: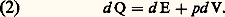 L’étude des transformations qui s’effectuent à volume V constant, appelées aussi isomètres ou encore isochores (d V = 0), permet d’introduire la notion de capacité thermique à volume constant sous la forme:
L’étude des transformations qui s’effectuent à volume V constant, appelées aussi isomètres ou encore isochores (d V = 0), permet d’introduire la notion de capacité thermique à volume constant sous la forme: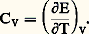 Lorsque la quantité de chaleur envisagée se rapporte à l’unité de masse ou à la mole, la capacité calorifique correspondante prend respectivement le nom de chaleur massique (anciennement chaleur spécifique ), et de chaleur molaire .On procède de même pour les transformations à pression constante appelées isobares (dp = 0). À cette fin, on introduit dans le principe de la conservation de l’énergie la fonction d’état H = E + p V, appelée enthalpie du système, ce qui permet d’écrire la relation (2) sous la forme:
Lorsque la quantité de chaleur envisagée se rapporte à l’unité de masse ou à la mole, la capacité calorifique correspondante prend respectivement le nom de chaleur massique (anciennement chaleur spécifique ), et de chaleur molaire .On procède de même pour les transformations à pression constante appelées isobares (dp = 0). À cette fin, on introduit dans le principe de la conservation de l’énergie la fonction d’état H = E + p V, appelée enthalpie du système, ce qui permet d’écrire la relation (2) sous la forme: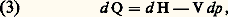 d’où la notion de capacité thermique à pression constante:
d’où la notion de capacité thermique à pression constante: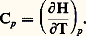 Les capacités thermiques Cp et CV sont donc aussi des fonctions d’état. Elles dépendent en général de la température, de la pression et de la composition du système. Pour un gaz parfait à un seul constituant, Cp et CV ne dépendent que de la température (loi de Joule). Au point de vue macroscopique, un gaz parfait est défini comme un système (p , V) obéissant aux lois de Joule et de Mariotte (loi de Boyle, 1662; loi de Mariotte, 1676): À température constante , le volume d’une masse donnée d’un gaz parfait varie en raison inverse de sa pression: p V = constante.Une transformation à température constante porte le nom d’isotherme. L’isotherme d’un gaz parfait est donc représentée par un arc d’hyperbole équilatère dans un système d’axes (p , V).Une évolution fréquemment considérée aussi est celle de l’adiabatique , accomplie par un système thermiquement isolé (d Q = 0). On déduit du principe de la conservation de l’énergie que l’évolution adiabatique d’un gaz parfait obéit à la loi de Laplace (P. S. Laplace, 1749-1827):
Les capacités thermiques Cp et CV sont donc aussi des fonctions d’état. Elles dépendent en général de la température, de la pression et de la composition du système. Pour un gaz parfait à un seul constituant, Cp et CV ne dépendent que de la température (loi de Joule). Au point de vue macroscopique, un gaz parfait est défini comme un système (p , V) obéissant aux lois de Joule et de Mariotte (loi de Boyle, 1662; loi de Mariotte, 1676): À température constante , le volume d’une masse donnée d’un gaz parfait varie en raison inverse de sa pression: p V = constante.Une transformation à température constante porte le nom d’isotherme. L’isotherme d’un gaz parfait est donc représentée par un arc d’hyperbole équilatère dans un système d’axes (p , V).Une évolution fréquemment considérée aussi est celle de l’adiabatique , accomplie par un système thermiquement isolé (d Q = 0). On déduit du principe de la conservation de l’énergie que l’évolution adiabatique d’un gaz parfait obéit à la loi de Laplace (P. S. Laplace, 1749-1827):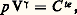 où 塚 = Cp /CV. Elle est valable dans tout le domaine où le rapport 塚 peut être considéré comme constant. La transformation isothermique est souvent utilisée comme le modèle d’une évolution infiniment lente, tandis que la transformation adiabatique représente au contraire le modèle d’une évolution quasi instantanée. L’introduction de la détente adiabatique dans le cycle de la machine à vapeur est due au physicien écossais James Watt (1736-1819). Elle a largement contribué au progrès ultérieur de la théorie mécanique de la chaleur. Les expressions «adiabatique» et «thermodynamique» sont attribuées à W. J. M. Rankine (1820-1872).À titre d’exemple, on a représenté sur la figure 1 un cycle de gaz parfait, composé de deux adiabatiques et de deux isothermes dans les axes (p , V), appelé cycle de Carnot. Ses propriétés jouent un rôle important en rapport avec le second principe de la thermodynamique considéré au chapitre 2. Le diagramme (p , V) porte généralement le nom de diagramme de Clapeyron (Émile Clapeyron, 1799-1864).Il est souvent utile de formuler la loi fondamentale (1) sous la forme d’un bilan d’énergie régissant les échanges avec le milieu extérieur. On écrit alors que l’accroissement d’énergie d U du système est attribuable, pour une part, à un flux d e U fourni par le milieu extérieur et, pour une seconde part, à une source provenant des phénomènes internes.Le bilan s’écrit donc:
où 塚 = Cp /CV. Elle est valable dans tout le domaine où le rapport 塚 peut être considéré comme constant. La transformation isothermique est souvent utilisée comme le modèle d’une évolution infiniment lente, tandis que la transformation adiabatique représente au contraire le modèle d’une évolution quasi instantanée. L’introduction de la détente adiabatique dans le cycle de la machine à vapeur est due au physicien écossais James Watt (1736-1819). Elle a largement contribué au progrès ultérieur de la théorie mécanique de la chaleur. Les expressions «adiabatique» et «thermodynamique» sont attribuées à W. J. M. Rankine (1820-1872).À titre d’exemple, on a représenté sur la figure 1 un cycle de gaz parfait, composé de deux adiabatiques et de deux isothermes dans les axes (p , V), appelé cycle de Carnot. Ses propriétés jouent un rôle important en rapport avec le second principe de la thermodynamique considéré au chapitre 2. Le diagramme (p , V) porte généralement le nom de diagramme de Clapeyron (Émile Clapeyron, 1799-1864).Il est souvent utile de formuler la loi fondamentale (1) sous la forme d’un bilan d’énergie régissant les échanges avec le milieu extérieur. On écrit alors que l’accroissement d’énergie d U du système est attribuable, pour une part, à un flux d e U fourni par le milieu extérieur et, pour une seconde part, à une source provenant des phénomènes internes.Le bilan s’écrit donc: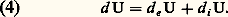 En particulier, pour un système fermé, la contribution extérieure est donnée par:
En particulier, pour un système fermé, la contribution extérieure est donnée par: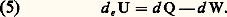 D’autre part, la source engendrée par les contributions internes doit être nulle, d’où:
D’autre part, la source engendrée par les contributions internes doit être nulle, d’où: En effet, dans le langage des bilans, le principe de la conservation de l’énergie revient à dire que la source d’énergie est identiquement nulle.La formulation (4) présente un intérêt particulier pour l’application des principes fondamentaux aux systèmes ouverts.2. Le second principeLe second principe de la thermodynamique trouve son origine dans le célèbre mémoire de Sadi Carnot intitulé Réflexions sur la puissance motrice du feu et sur les machines propres à développer cette puissance , publié à Paris, en 1824 [cf. CARNOT (S.)].Toutefois, comme il s’y trouvait exposé dans l’ancien langage du calorique, il a fallu toute la perspicacité de E. Clapeyron et de W. Thomson (lord Kelvin) pour le redécouvrir dans l’œuvre originale, au cours des années 1834 pour le premier, et 1848-1849 pour le second [cf. KELVIN (lord)].Développant quelques considérations relatives aux cycles, établies par Carnot à partir de l’impossibilité du mouvement perpétuel de seconde espèce (il est impossible de décrire un cycle direct à l’aide d’une seule source de chaleur), lord Kelvin établit l’échelle thermodynamique absolue de température T qui porte aujourd’hui son nom. Il prouve en même temps l’existence du zéro absolu, le même pour tous les corps (face=F0019 漣 273,15 0C), et obtient pour le gaz parfait (cf. MESURE -mesures thermiques) l’équation caractéristique simple:
En effet, dans le langage des bilans, le principe de la conservation de l’énergie revient à dire que la source d’énergie est identiquement nulle.La formulation (4) présente un intérêt particulier pour l’application des principes fondamentaux aux systèmes ouverts.2. Le second principeLe second principe de la thermodynamique trouve son origine dans le célèbre mémoire de Sadi Carnot intitulé Réflexions sur la puissance motrice du feu et sur les machines propres à développer cette puissance , publié à Paris, en 1824 [cf. CARNOT (S.)].Toutefois, comme il s’y trouvait exposé dans l’ancien langage du calorique, il a fallu toute la perspicacité de E. Clapeyron et de W. Thomson (lord Kelvin) pour le redécouvrir dans l’œuvre originale, au cours des années 1834 pour le premier, et 1848-1849 pour le second [cf. KELVIN (lord)].Développant quelques considérations relatives aux cycles, établies par Carnot à partir de l’impossibilité du mouvement perpétuel de seconde espèce (il est impossible de décrire un cycle direct à l’aide d’une seule source de chaleur), lord Kelvin établit l’échelle thermodynamique absolue de température T qui porte aujourd’hui son nom. Il prouve en même temps l’existence du zéro absolu, le même pour tous les corps (face=F0019 漣 273,15 0C), et obtient pour le gaz parfait (cf. MESURE -mesures thermiques) l’équation caractéristique simple: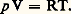 Partant de ces résultats, Clausius en a déduit, en 1850, une formulation mathématique du second principe si générale que, depuis cette époque, on lui a donné le nom de principe de Carnot-Clausius. Il montre d’abord que, pour l’ensemble des processus réversibles , la température T est un diviseur intégrant de d Q et que, par conséquent, on peut écrire, pour un système fermé à température uniforme, une égalité du type suivant:
Partant de ces résultats, Clausius en a déduit, en 1850, une formulation mathématique du second principe si générale que, depuis cette époque, on lui a donné le nom de principe de Carnot-Clausius. Il montre d’abord que, pour l’ensemble des processus réversibles , la température T est un diviseur intégrant de d Q et que, par conséquent, on peut écrire, pour un système fermé à température uniforme, une égalité du type suivant: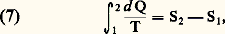 où S2 漣 S1 désigne l’accroissement d’une fonction d’état du système dénommée entropie (en grec évolution ). Le raisonnement suivi est le même que pour l’introduction de l’énergie comme fonction d’état dans l’équation (1). Dans les deux cas, la grandeur introduite n’est définie que par ses accroissements, c’est-à-dire à une constante près.Portant ensuite son attention sur les transformations irréversibles d’un système fermé et utilisant le résultat ci-dessus Clausius en déduit l’inégalité fondamentale suivante des processus irréversibles :
où S2 漣 S1 désigne l’accroissement d’une fonction d’état du système dénommée entropie (en grec évolution ). Le raisonnement suivi est le même que pour l’introduction de l’énergie comme fonction d’état dans l’équation (1). Dans les deux cas, la grandeur introduite n’est définie que par ses accroissements, c’est-à-dire à une constante près.Portant ensuite son attention sur les transformations irréversibles d’un système fermé et utilisant le résultat ci-dessus Clausius en déduit l’inégalité fondamentale suivante des processus irréversibles :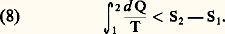 Pour obtenir cette dernière relation, il doit cependant admettre l’existence d’au moins une transformation réversible permettant de relier l’état initial et l’état final. Enfin, pour une transformation infinitésimale, il déduit directement des relations (7) et (8) l’inégalité différentielle, aujourd’hui classique:
Pour obtenir cette dernière relation, il doit cependant admettre l’existence d’au moins une transformation réversible permettant de relier l’état initial et l’état final. Enfin, pour une transformation infinitésimale, il déduit directement des relations (7) et (8) l’inégalité différentielle, aujourd’hui classique: où le signe d’égalité correspond à une transformation élémentaire réversible, c’est-à-dire à un déplacement d’équilibre.En vue de l’extension des relations ci-dessus aux systèmes ouverts, il est commode d’exprimer le second principe sous la forme d’un bilan. Parallèlement au bilan d’énergie (4), on écrit le bilan entropique sous la forme [cf. IRRÉVERSIBILITÉ]:
où le signe d’égalité correspond à une transformation élémentaire réversible, c’est-à-dire à un déplacement d’équilibre.En vue de l’extension des relations ci-dessus aux systèmes ouverts, il est commode d’exprimer le second principe sous la forme d’un bilan. Parallèlement au bilan d’énergie (4), on écrit le bilan entropique sous la forme [cf. IRRÉVERSIBILITÉ]: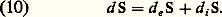 Le terme d e S représente la contribution du milieu extérieur à l’accroissement d’entropie; c’est le flux entropique reçu par le système. Il se réduit à d Q/T pour un système fermé à température uniforme. Quant au terme de source d i S il correspond à l’accroissement d’entropie engendré par les processus irréversibles internes.Le second principe impose un signe non négatif à cette source. Rapportée à l’unité de temps, on l’appelle généralement la production d’entropie :
Le terme d e S représente la contribution du milieu extérieur à l’accroissement d’entropie; c’est le flux entropique reçu par le système. Il se réduit à d Q/T pour un système fermé à température uniforme. Quant au terme de source d i S il correspond à l’accroissement d’entropie engendré par les processus irréversibles internes.Le second principe impose un signe non négatif à cette source. Rapportée à l’unité de temps, on l’appelle généralement la production d’entropie : Pour un système isolé, l’état d’équilibre correspond à un maximum de l’entropie. Toutefois, dans tous les cas, il correspond au minimum de la production d’entropie. Comme on le constatera plus loin, les états stationnaires de non-équilibre relevant du domaine de la thermodynamique linéaire possèdent cette même dernière propriété, mais alors le minimum n’est plus nul.Dans le cas simple du milieu homogène dont l’état est entièrement défini par deux variables indépendantes, la condition de réversibilité est supposée toujours satisfaite. On obtient ainsi, à partir des expressions (2) et (3) et de l’égalité figurant dans (9), les relations:
Pour un système isolé, l’état d’équilibre correspond à un maximum de l’entropie. Toutefois, dans tous les cas, il correspond au minimum de la production d’entropie. Comme on le constatera plus loin, les états stationnaires de non-équilibre relevant du domaine de la thermodynamique linéaire possèdent cette même dernière propriété, mais alors le minimum n’est plus nul.Dans le cas simple du milieu homogène dont l’état est entièrement défini par deux variables indépendantes, la condition de réversibilité est supposée toujours satisfaite. On obtient ainsi, à partir des expressions (2) et (3) et de l’égalité figurant dans (9), les relations: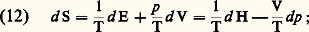 soit, par exemple, pour un gaz parfait (p V = RT):
soit, par exemple, pour un gaz parfait (p V = RT):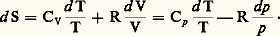 Ces deux dernières relations servent de base à la construction du diagramme entropique (T,S) des gaz parfaits [cf. ENTROPIE].Pour un grand nombre d’évolutions accompagnées d’effets dissipatifs, l’état local peut être décrit à l’aide des mêmes variables que l’état d’équilibre. Dans ces conditions, on maintient les égalités (12) écrites sous la forme locale comme expression de l’entropie spécifique (les variables spécifiques sont représentées par les minuscules correspondantes et se rapportent à l’unité de masse):
Ces deux dernières relations servent de base à la construction du diagramme entropique (T,S) des gaz parfaits [cf. ENTROPIE].Pour un grand nombre d’évolutions accompagnées d’effets dissipatifs, l’état local peut être décrit à l’aide des mêmes variables que l’état d’équilibre. Dans ces conditions, on maintient les égalités (12) écrites sous la forme locale comme expression de l’entropie spécifique (les variables spécifiques sont représentées par les minuscules correspondantes et se rapportent à l’unité de masse):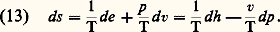 Ces relations traduisent l’hypothèse de l’équilibre local. Plus généralement, lorsque des changements massiques peuvent intervenir à côté des changements de température et de pression, la formule locale explicite de l’entropie est donnée par l’équation de Gibbs :
Ces relations traduisent l’hypothèse de l’équilibre local. Plus généralement, lorsque des changements massiques peuvent intervenir à côté des changements de température et de pression, la formule locale explicite de l’entropie est donnée par l’équation de Gibbs :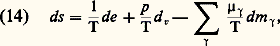 Cette importante relation, qui se trouve à la base de l’étude des phénomènes physico-chimiques, a été établie dans le célèbre mémoire Equilibrium of Heterogeneous Substances (1874-1878). Dans cette relation, les changements de masses m size=1塚 des différents constituants 塚 sont arbitraires. Ils proviennent soit de phénomènes physico-chimiques internes, soit d’échanges de matière avec le milieu extérieur (systèmes ouverts). D’autre part, les coefficients 猪 size=1塚 représentent les potentiels chimiques massiques de ces constituants (cf. équation 18).Dans tous les problèmes pour lesquels la formule de Gibbs est utilisable, la production d’entropie locale 靖 devient une forme bilinéaire des courants généralisés Ji , ainsi que des forces généralisées Xi , servant à caractériser les causes de dissipation d’énergie. On obtient ainsi, pour un système de volume V, l’expression globale [cf. IRRÉVERSIBILITÉ]:
Cette importante relation, qui se trouve à la base de l’étude des phénomènes physico-chimiques, a été établie dans le célèbre mémoire Equilibrium of Heterogeneous Substances (1874-1878). Dans cette relation, les changements de masses m size=1塚 des différents constituants 塚 sont arbitraires. Ils proviennent soit de phénomènes physico-chimiques internes, soit d’échanges de matière avec le milieu extérieur (systèmes ouverts). D’autre part, les coefficients 猪 size=1塚 représentent les potentiels chimiques massiques de ces constituants (cf. équation 18).Dans tous les problèmes pour lesquels la formule de Gibbs est utilisable, la production d’entropie locale 靖 devient une forme bilinéaire des courants généralisés Ji , ainsi que des forces généralisées Xi , servant à caractériser les causes de dissipation d’énergie. On obtient ainsi, pour un système de volume V, l’expression globale [cf. IRRÉVERSIBILITÉ]: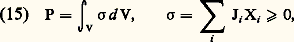 où 靖 désigne la production d’entropie volumique. L’inégalité représente une contrainte imposée aux courants, en sorte que la condition (15) s’interprète comme un critère d’évolution.En particulier, dans le problème de la conduction thermique, le courant de chaleur est donné par la loi vectorielle de Fourier J = 漣暴T (cf. CHALEUR, chap. 5). D’où la production d’entropie correspondante:
où 靖 désigne la production d’entropie volumique. L’inégalité représente une contrainte imposée aux courants, en sorte que la condition (15) s’interprète comme un critère d’évolution.En particulier, dans le problème de la conduction thermique, le courant de chaleur est donné par la loi vectorielle de Fourier J = 漣暴T (cf. CHALEUR, chap. 5). D’où la production d’entropie correspondante: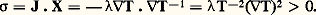 De même, lorsque les masses m size=1塚 sont engagées dans une réaction chimique du type:
De même, lorsque les masses m size=1塚 sont engagées dans une réaction chimique du type: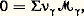 où les 益 size=1塚 sont les coefficients et où les 紐 塚 sont les masses molaires des constituants ( 塚 = 1 à c ), la contribution chimique à la production d’entropie conduit à la forme bilinéaire:
où les 益 size=1塚 sont les coefficients et où les 紐 塚 sont les masses molaires des constituants ( 塚 = 1 à c ), la contribution chimique à la production d’entropie conduit à la forme bilinéaire: où la grandeur A désigne l’affinité chimique de la réaction et où u est la vitesse de réaction correspondante.Cette affinité s’exprime, en fonction des potentiels chimiques, à l’aide de l’égalité:
où la grandeur A désigne l’affinité chimique de la réaction et où u est la vitesse de réaction correspondante.Cette affinité s’exprime, en fonction des potentiels chimiques, à l’aide de l’égalité: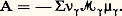 On a, de plus, u = d 﨡/dt , où 﨡 désigne le degré d’avancement de la réaction , ou encore la variable chimique. On a pour un système fermé:
On a, de plus, u = d 﨡/dt , où 﨡 désigne le degré d’avancement de la réaction , ou encore la variable chimique. On a pour un système fermé: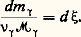 L’inégalité (16) et la notion d’affinité associée, conçue comme une fonction d’état et non comme une simple réaction à une résistance de frottement chimique au sens envisagé antérieurement par Duhem, ont été introduites, dès 1922, par T. De Donder.Il faut oberver que, chaque fois qu’on peut invoquer l’hypothèse de l’équilibre local, la restriction citée antérieurement concernant l’existence d’au moins une transformation réversible entre deux états donnés est sans importance. En effet, l’entropie se trouve alors définie par la formule de Gibbs en chaque point du système.Mais l’hypothèse de l’équilibre local n’est pas acceptable dans tous les cas. La rhéologie, par exemple, offre de nombreux problèmes pour lesquels la formule de Gibbs n’est pas applicable, même localement, soit que de nouvelles variables indépendantes doivent être introduites, soit que la description de l’état local implique la connaissance des gradients. Il est alors généralement nécessaire de recourir à la définition extra-thermodynamique de l’entropie, introduite par Boltzmann:
L’inégalité (16) et la notion d’affinité associée, conçue comme une fonction d’état et non comme une simple réaction à une résistance de frottement chimique au sens envisagé antérieurement par Duhem, ont été introduites, dès 1922, par T. De Donder.Il faut oberver que, chaque fois qu’on peut invoquer l’hypothèse de l’équilibre local, la restriction citée antérieurement concernant l’existence d’au moins une transformation réversible entre deux états donnés est sans importance. En effet, l’entropie se trouve alors définie par la formule de Gibbs en chaque point du système.Mais l’hypothèse de l’équilibre local n’est pas acceptable dans tous les cas. La rhéologie, par exemple, offre de nombreux problèmes pour lesquels la formule de Gibbs n’est pas applicable, même localement, soit que de nouvelles variables indépendantes doivent être introduites, soit que la description de l’état local implique la connaissance des gradients. Il est alors généralement nécessaire de recourir à la définition extra-thermodynamique de l’entropie, introduite par Boltzmann: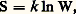 où W est la probabilité de l’état considéré (cf. ENTROPIE, théorie de l’INFORMATION, IRRÉVERSIBILITÉ, mécanique STATISTIQUE) et servant à mesurer le degré de désordre du système.3. Les potentiels thermodynamiques, les états d’équilibre et la stabilité de l’équilibreL’élimination de la différentielle non exacte d Q entre les expressions données plus haut du premier et du second principe conduit aux relations:
où W est la probabilité de l’état considéré (cf. ENTROPIE, théorie de l’INFORMATION, IRRÉVERSIBILITÉ, mécanique STATISTIQUE) et servant à mesurer le degré de désordre du système.3. Les potentiels thermodynamiques, les états d’équilibre et la stabilité de l’équilibreL’élimination de la différentielle non exacte d Q entre les expressions données plus haut du premier et du second principe conduit aux relations: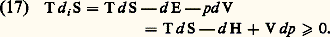 On en déduit les critères d’évolution suivants: pour un système maintenu à V et à S constants, l’énergie E décroît (d E 諒 0) et il en est de même à p et à S constants pour l’enthalpie (d H 諒 0). Le signe d’égalité correspond à l’état d’équilibre.Comme l’emploi des variables (V, T) ou (p ,T) est beaucoup plus commode en pratique que celui des variables (V, S) ou (p , S), on opère un changement de variables sur les relations précédentes. En posant:
On en déduit les critères d’évolution suivants: pour un système maintenu à V et à S constants, l’énergie E décroît (d E 諒 0) et il en est de même à p et à S constants pour l’enthalpie (d H 諒 0). Le signe d’égalité correspond à l’état d’équilibre.Comme l’emploi des variables (V, T) ou (p ,T) est beaucoup plus commode en pratique que celui des variables (V, S) ou (p , S), on opère un changement de variables sur les relations précédentes. En posant: on obtient comme critère d’évolution la décroissance de F, à température T et à volume V constants (d F 諒 0), ou celle de G, à température T et à pression p constante (d G 諒 0). On a donné le nom de potentiel thermodynamique à ces deux nouvelles fonctions d’état F et G. On les appelle aussi fonctions de Massieu. Toutefois, les expressions les plus employées actuellement sont celles d’énergie libre de Helmholtz pour F et d’énergie libre de Gibbs pour G, dite aussi enthalpie libre .Le potentiel chimique 猪 size=1塚 utilisé plus haut est lié aux grandeurs ci-dessus par les égalités:
on obtient comme critère d’évolution la décroissance de F, à température T et à volume V constants (d F 諒 0), ou celle de G, à température T et à pression p constante (d G 諒 0). On a donné le nom de potentiel thermodynamique à ces deux nouvelles fonctions d’état F et G. On les appelle aussi fonctions de Massieu. Toutefois, les expressions les plus employées actuellement sont celles d’énergie libre de Helmholtz pour F et d’énergie libre de Gibbs pour G, dite aussi enthalpie libre .Le potentiel chimique 猪 size=1塚 utilisé plus haut est lié aux grandeurs ci-dessus par les égalités: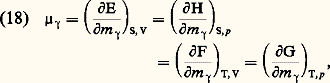 et l’on a:
et l’on a: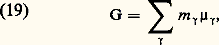 avec 塚 = 1, ..., c .Quelques autres relations thermodynamiques fondamentales font appel aux propriétés de l’une ou de l’autre de ces énergies libres. On se contentera ici de signaler les formules de Gibbs-Helmholtz :
avec 塚 = 1, ..., c .Quelques autres relations thermodynamiques fondamentales font appel aux propriétés de l’une ou de l’autre de ces énergies libres. On se contentera ici de signaler les formules de Gibbs-Helmholtz :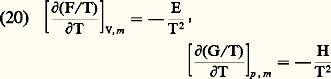 et la formule de Gibbs-Duhem :
et la formule de Gibbs-Duhem :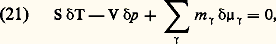 pour toute variation 嗀, réelle ou virtuelle, des variables T, p et 猪 size=1塚.Quant aux états d’équilibre, ils sont caractérisés par un extremum de F, à T et à V constants, ou de G, à T et à p constants. Recherchons, à titre d’exemple, la condition d’équilibre d’un système à deux phases et à un constituant. L’extremum de G, à T et à p constants, s’écrit:
pour toute variation 嗀, réelle ou virtuelle, des variables T, p et 猪 size=1塚.Quant aux états d’équilibre, ils sont caractérisés par un extremum de F, à T et à V constants, ou de G, à T et à p constants. Recherchons, à titre d’exemple, la condition d’équilibre d’un système à deux phases et à un constituant. L’extremum de G, à T et à p constants, s’écrit: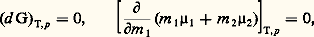
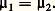 Comme le potentiel G doit être nécessairement une fonction linéaire et homogène des masses, les potentiels chimiques 猪 size=1塚 apparaissent comme des fonctions de degré zéro. Dans le cas d’un corps pur, les 猪 size=1塚 sont donc des fonctions de la température et de la pression uniquement, et l’égalité ci-dessus exprime simplement une condition de la forme f (T, p ) = 0 pour l’équilibre des deux phases, qu’on appelle ordinairement courbe des tensions de vapeur.Le même type de raisonnement conduit à l’étude des déplacements d’équilibre. À cette fin, on écrit la condition d’équilibre 猪1 = 猪2 entre deux états infiniment voisins:
Comme le potentiel G doit être nécessairement une fonction linéaire et homogène des masses, les potentiels chimiques 猪 size=1塚 apparaissent comme des fonctions de degré zéro. Dans le cas d’un corps pur, les 猪 size=1塚 sont donc des fonctions de la température et de la pression uniquement, et l’égalité ci-dessus exprime simplement une condition de la forme f (T, p ) = 0 pour l’équilibre des deux phases, qu’on appelle ordinairement courbe des tensions de vapeur.Le même type de raisonnement conduit à l’étude des déplacements d’équilibre. À cette fin, on écrit la condition d’équilibre 猪1 = 猪2 entre deux états infiniment voisins: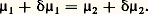 Il en résulte 嗀猪1 = 嗀猪2, d’où, d’après l’expression différentielle de l’énergie libre de Gibbs:
Il en résulte 嗀猪1 = 嗀猪2, d’où, d’après l’expression différentielle de l’énergie libre de Gibbs: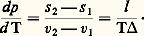 Cette expression donne la direction de la tangente en un point de la courbe des tensions de vapeur lorsque l’une des phases est gazeuse. C’est la loi de Clapeyron . On notera que l représente la chaleur de la transition de phase et , la dilatation correspondante. En outre, on observera que cette formule permet la détermination de la température absolue sans avoir à recourir aux propriétés des gaz parfaits.Semblablement, l’équation de Schröder-van Laar , qui régit les courbes de cristallisation de solutions idéales conduisant à la formation d’eutectiques, s’obtient sous la forme connue:
Cette expression donne la direction de la tangente en un point de la courbe des tensions de vapeur lorsque l’une des phases est gazeuse. C’est la loi de Clapeyron . On notera que l représente la chaleur de la transition de phase et , la dilatation correspondante. En outre, on observera que cette formule permet la détermination de la température absolue sans avoir à recourir aux propriétés des gaz parfaits.Semblablement, l’équation de Schröder-van Laar , qui régit les courbes de cristallisation de solutions idéales conduisant à la formation d’eutectiques, s’obtient sous la forme connue: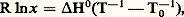 où x désigne la fraction molaire du constituant en solution, et H0 sa chaleur latente de fusion à la température 0 de congélation du constituant pur.Toute la théorie de l’équilibre et du déplacement d’équilibre des systèmes à plusieurs constituants se déduit de la même façon dans le cadre de la thermodynamique chimique. Elle comporte notamment la règle des phases sur la variance d’un système physico-chimique en équilibre, établie par J. W. Gibbs dans le mémoire cité plus haut (cf. THERMODYNAMIQUE – Thermodynamique chimique).D’autre part, les inégalités (17) fournissent également des critères de stabilité de l’état d’équilibre selon une méthode introduite par J. W. Gibbs et P. Duhem. On exprime à l’aide de (17) qu’aucune évolution partant de l’état d’équilibre n’est possible parce qu’elle ne respecterait pas le signe d’inégalité imposé par le second principe aux processus naturels. Utilisant le symbole 嗀 pour désigner un déplacement virtuel, la condition de stabilité de l’équilibre s’écrit sous l’une des formes:
où x désigne la fraction molaire du constituant en solution, et H0 sa chaleur latente de fusion à la température 0 de congélation du constituant pur.Toute la théorie de l’équilibre et du déplacement d’équilibre des systèmes à plusieurs constituants se déduit de la même façon dans le cadre de la thermodynamique chimique. Elle comporte notamment la règle des phases sur la variance d’un système physico-chimique en équilibre, établie par J. W. Gibbs dans le mémoire cité plus haut (cf. THERMODYNAMIQUE – Thermodynamique chimique).D’autre part, les inégalités (17) fournissent également des critères de stabilité de l’état d’équilibre selon une méthode introduite par J. W. Gibbs et P. Duhem. On exprime à l’aide de (17) qu’aucune évolution partant de l’état d’équilibre n’est possible parce qu’elle ne respecterait pas le signe d’inégalité imposé par le second principe aux processus naturels. Utilisant le symbole 嗀 pour désigner un déplacement virtuel, la condition de stabilité de l’équilibre s’écrit sous l’une des formes: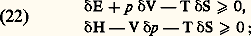 d’où les cas particuliers suivants: pour (V, S) ou (p , S) constants, un état d’équilibre stable correspond au minimum de l’énergie interne E (ou de l’enthalpie H). De même, en variable (T, V) ou (T, p ), c’est l’énergie libre F (ou l’enthalpie libre G) qui doit être minimale. Ces différents critères conduisent aux conditions explicites de stabilité de l’équilibre suivantes: CV 礪 0 (stabilité thermique) et (face=F0019 煉V/ 煉p )T 麗 0 (stabilité mécanique). De plus, la condition de stabilité de diffusion, qui comporte la stabilité de l’équilibre chimique, s’écrit (signe sommatoire sous-entendu):
d’où les cas particuliers suivants: pour (V, S) ou (p , S) constants, un état d’équilibre stable correspond au minimum de l’énergie interne E (ou de l’enthalpie H). De même, en variable (T, V) ou (T, p ), c’est l’énergie libre F (ou l’enthalpie libre G) qui doit être minimale. Ces différents critères conduisent aux conditions explicites de stabilité de l’équilibre suivantes: CV 礪 0 (stabilité thermique) et (face=F0019 煉V/ 煉p )T 麗 0 (stabilité mécanique). De plus, la condition de stabilité de diffusion, qui comporte la stabilité de l’équilibre chimique, s’écrit (signe sommatoire sous-entendu):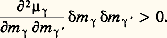 Comme cette expression est une forme quadratique définie positive, les conditions explicites de stabilité sont données par les signes imposés aux coefficients.Par exemple, l’arc m M de l’isotherme de Van der Waals (J. D. Van der Waals, 1837-1923) représentée sur la figure 2 (cf. état GAZEUX, chap. 3) correspond à une suite d’états instables puisqu’on a dans ce cas:
Comme cette expression est une forme quadratique définie positive, les conditions explicites de stabilité sont données par les signes imposés aux coefficients.Par exemple, l’arc m M de l’isotherme de Van der Waals (J. D. Van der Waals, 1837-1923) représentée sur la figure 2 (cf. état GAZEUX, chap. 3) correspond à une suite d’états instables puisqu’on a dans ce cas: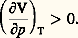 Cette instabilité interdit le maintien d’un état gazeux homogène le long de l’arc m M, ce qui donne naissance à la condensation. Le système tend alors vers un nouvel état d’équilibre non homogène, se présentant sous deux phases, et satisfaisant à la condition exprimée par la courbe des tensions de vapeur f (T, p ) = 0, citée plus haut (segment horizontal LV sur la figure 2).L’entropie n’ayant été définie qu’à une constante près, le calcul de l’accroissement F ou G de l’énergie libre d’un composé quelconque exige soit la connaissance du lien entre les constantes d’entropie des différents constituants, soit celle de la valeur absolue de chaque entropie. Le calcul est largement facilité grâce à l’intervention d’une hypothèse supplémentaire qui porte le nom d’hypothèse de Nernst (W. Nernst, 1864-1941), appelée souvent, mais abusivement, troisième principe de la thermodynamique (1906). On peut l’énoncer comme suit: «L’entropie d’un corps pur cristallisé est la même pour tous les corps au zéro absolu de température. Elle est indépendante de la pression et des modifications chimiques du système.» Comme la valeur constante qu’on peut lui attribuer disparaît des calculs, M. Planck (1858-1947) a proposé de la prendre nulle, ce qui conduit à l’énoncé simplifié mieux connu: «Les corps cristallisés ont une entropie nulle au zéro absolu.» De là, on déterminera par un calcul direct l’entropie du solide aux températures successives, ensuite celles du liquide et de l’état gazeux, à l’aide des données calorimétriques relatives aux chaleurs massiques du solide, du liquide et du gaz, ainsi qu’aux chaleurs de transitions de phase (fusion et évaporation).
Cette instabilité interdit le maintien d’un état gazeux homogène le long de l’arc m M, ce qui donne naissance à la condensation. Le système tend alors vers un nouvel état d’équilibre non homogène, se présentant sous deux phases, et satisfaisant à la condition exprimée par la courbe des tensions de vapeur f (T, p ) = 0, citée plus haut (segment horizontal LV sur la figure 2).L’entropie n’ayant été définie qu’à une constante près, le calcul de l’accroissement F ou G de l’énergie libre d’un composé quelconque exige soit la connaissance du lien entre les constantes d’entropie des différents constituants, soit celle de la valeur absolue de chaque entropie. Le calcul est largement facilité grâce à l’intervention d’une hypothèse supplémentaire qui porte le nom d’hypothèse de Nernst (W. Nernst, 1864-1941), appelée souvent, mais abusivement, troisième principe de la thermodynamique (1906). On peut l’énoncer comme suit: «L’entropie d’un corps pur cristallisé est la même pour tous les corps au zéro absolu de température. Elle est indépendante de la pression et des modifications chimiques du système.» Comme la valeur constante qu’on peut lui attribuer disparaît des calculs, M. Planck (1858-1947) a proposé de la prendre nulle, ce qui conduit à l’énoncé simplifié mieux connu: «Les corps cristallisés ont une entropie nulle au zéro absolu.» De là, on déterminera par un calcul direct l’entropie du solide aux températures successives, ensuite celles du liquide et de l’état gazeux, à l’aide des données calorimétriques relatives aux chaleurs massiques du solide, du liquide et du gaz, ainsi qu’aux chaleurs de transitions de phase (fusion et évaporation).
Encyclopédie Universelle. 2012.